Portfolio
- Research
- Open Source Software
- Freelance
-
Independent machine learning & AI projects
- Machine learning & AI APIs in production
- Survival analysis using gene expression & clinical data (Cox models)
- Deep learning for X-ray images and scRNAseq
- GNN for spatial transcriptomics
- LLM-based assistant for bioinformatics queries
- Computational biology & algorithms
- Protein folding - Monte Carlo
- Genome assembly - de Bruijn graph, euler walk
- Evolutionary tree estimation - Felsenstein & NNI
- Regulatory DNA discovery - MSA & binomial enrichment
- Games: Sudoku (JavaScript) and Minesweeper (Java)
- Django web services (Django-based Multiple Sequence Alignment visualization, Django-based mobile app, Pyodine-based in-browser Python career-matching tool)
Research
We used machine learning models - elastic net regression and principal component analysis (PCA) - to investigate genomic regions called 'HOT regions' which appear to attract unusually high numbers of proteins and are likely technical artifacts of chromatin immunoprecipitation followed by sequencing (ChIP-seq) experiments.
While factors like antibody quality and chromatin interactions are known to affect ChIP-seq reliability, our study revealed that GC- and CpG-rich sequences, DNA methylation, and RNA:DNA hybrids (R-loops) also contribute to these artifacts across species. This work shows how machine learning can uncover hidden biases in genomic data and improve experimental interpretation.
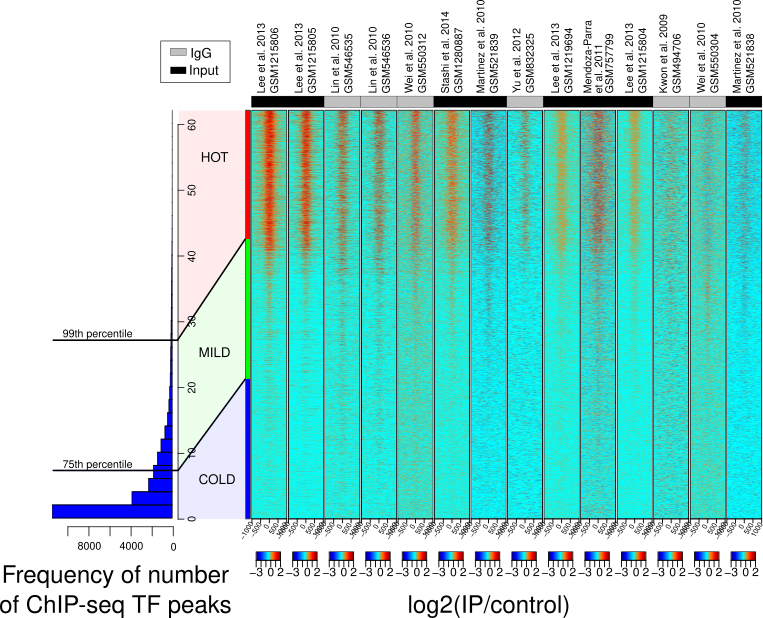
📄 Publication: Wreczycka K et al, Nucleic Acids Research, 2019
We explored circulating cell-free DNA (cfDNA) methylation as a non-invasive biomarker for acute coronary syndrome (ACS), based on the principle that damaged tissues release DNA into the bloodstream.
Using cfDNA methylation profiles, we differentiated ACS subtypes and identified cell type-specific DNA methylation markers to trace the origin of cfDNA. Hundreds of methylation markers linked to cardiovascular conditions and inflammation were identified and validated in an independent cohort, highlighting the potential of cfDNA methylation for ACS diagnosis.
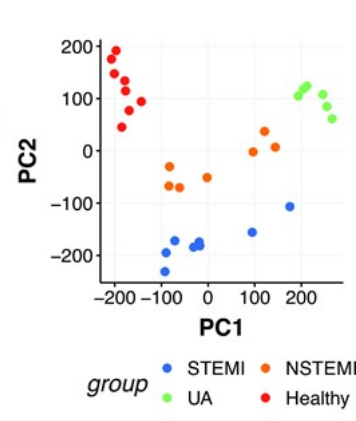
📄 Publication: Rafael R C Cuadrat et al, NAR Genomics and Bioinformatics, 2023
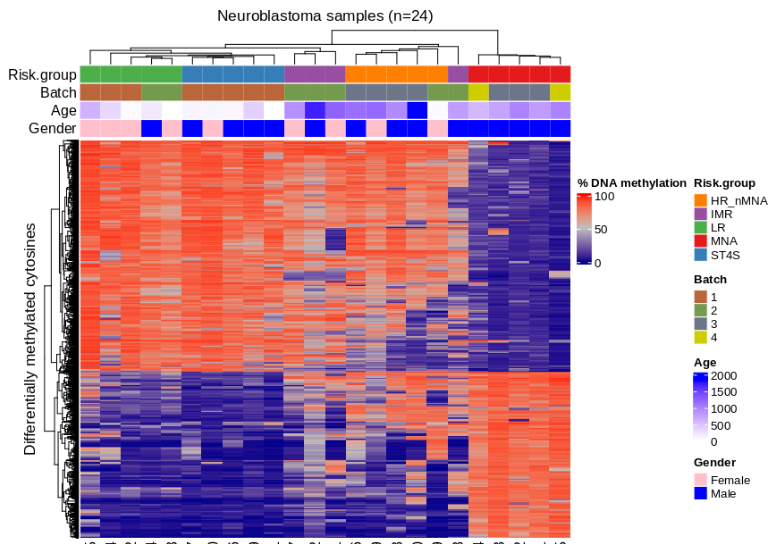
Neuroblastoma is a pediatric cancer ranging from mild to aggressive forms. While genetic changes explain some variability, we showed that DNA methylation plays a key role in its progression. In collaboration with Charité Hospital (Berlin), we analyzed primary tumor tissues and urine cfDNA using bisulfite-seq and RNA-seq, identifying methylation patterns distinguishing high- and low-risk tumors. We also linked MYCN-driven methylation changes to disrupted transcription factor networks, highlighting potential targets for therapies.
Open source software

genomation – a Bioconductor R package designed to simplify genomic feature and interval analysis. It includes functions for reading BED/GFF files as GRanges, summarizing features over regions, creating enrichment plots or heatmaps, and annotating regions with exons, introns, or promoters. I was a co-developer and maintainer.
https://github.com/BIMSBbioinfo/genomation, developed in the team of Dr. Altuna Akalin at Bioinformatics and Omics Data Science Platform at MDC BIMSB.

PiGx – a collection of genomics pipelines implemented using Snakemake, Python, and R. Each pipeline is easily configured with a sample sheet and a simple settings file. PiGx generates comprehensive, interactive HTML reports that summarize key findings from your samples. My contribution was co-implementing PigX BS-seq.
https://github.com/BIMSBbioinfo/pigx, developed in the team of Dr. Altuna Akalin at Bioinformatics and Omics Data Science Platform at MDC BIMSB.
motifActivity – an R package for identifying key transcription factors (TFs) responsible for changes in gene expression or epigenetic marks across samples. It predicts TF activity profiles using input data from RNA-seq, BS-seq, ChIP-seq, ATAC-seq, and similar methods, combined with a set of DNA motifs.
https://github.com/katwre/motifActivity, developed in the team of Dr. Altuna Akalin at Bioinformatics and Omics Data Science Platform at MDC BIMSB.
Freelance
We developed interactive visualizations, including oncoprints, to highlight key biomarkers in patients with limited treatment options. These visual summaries help uncover genomic alterations and support identifying new therapeutic targets.
We focused on patients from clinical trial databases facing poor outcomes or lacking effective therapies. Our statistical analyses, including survival analysis, demonstrate the clinical relevance of nominated targets.
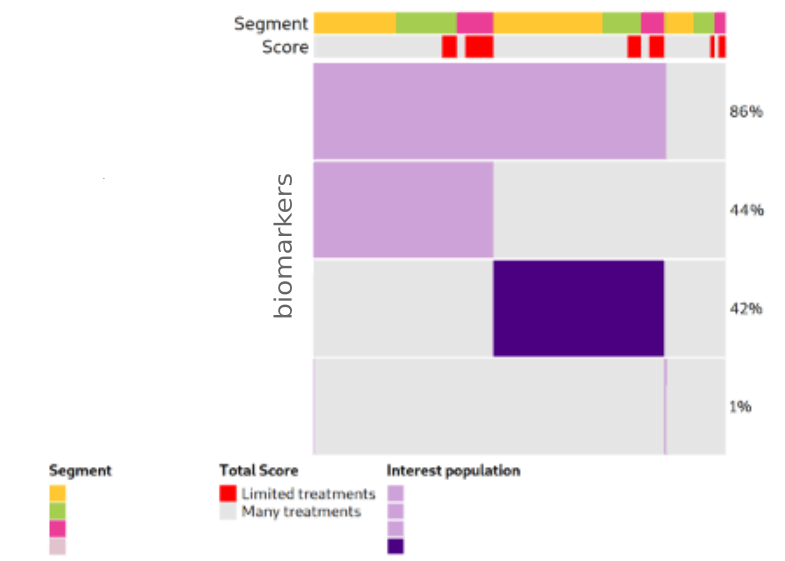
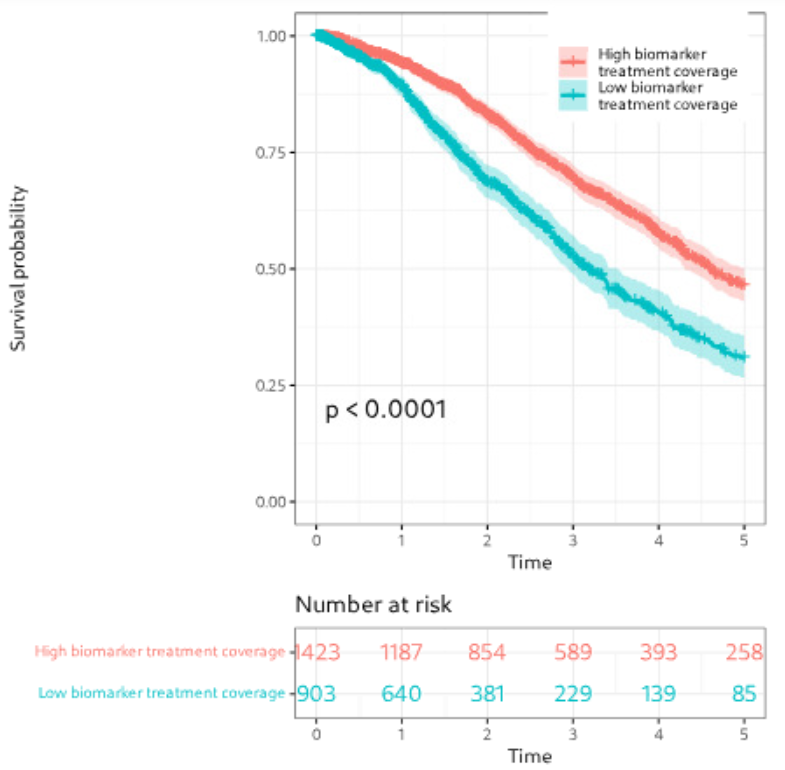
Figure: Example of biomarker visualization and survival analysis.
To prioritize therapeutic targets, we applied Positive and Unlabeled (PU) learning, ideal for cases where only confirmed targets are known. PU classifiers helped distinguish potential targets using gene expression, mutations, and therapy annotations.
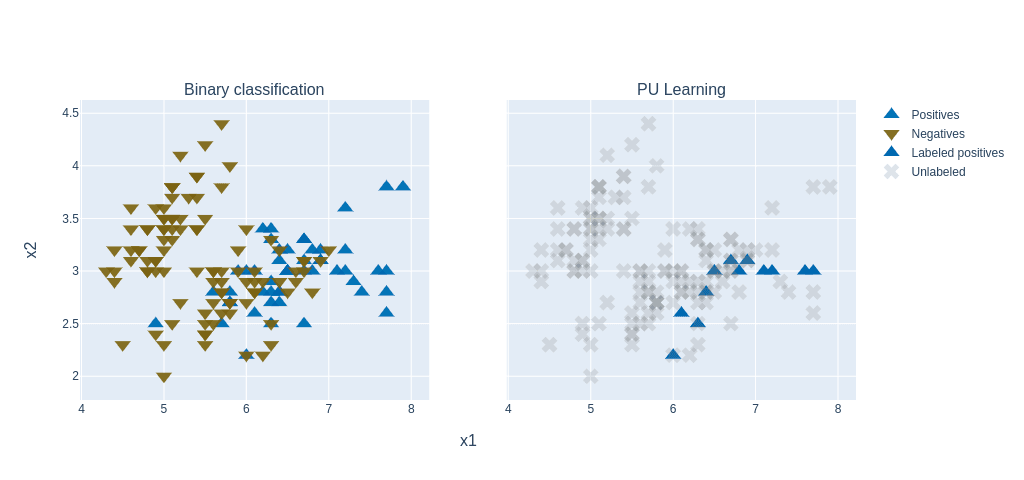
Additionally, we utilized autoencoders to uncover hidden patterns and prioritize key molecular features in an unsupervised way.
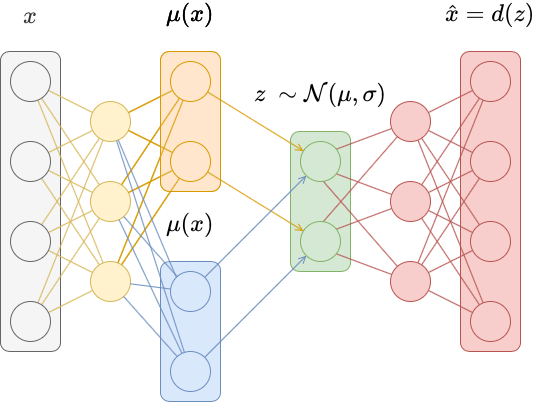
Multi-omics and AI (Enformer) for an Alzheimer's disease biomarker
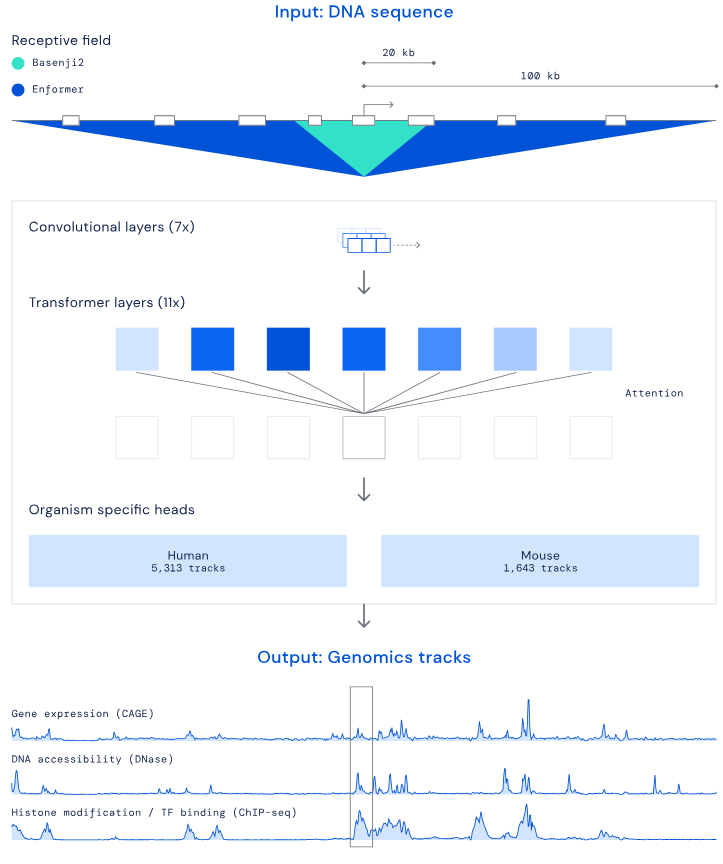
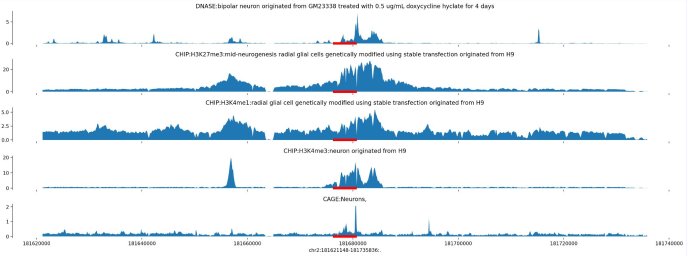
In this project, I investigated glial-to-neuron reprogramming through activation of a specific transcription factor (TF) in Alzheimer’s disease. I integrated multi-omics data - including RNA-seq, ATAC-seq, and H3K4me2/H3K27me3 ChIP-seq - to identify differentially expressed genes and gene pathways (GO, GSEA), their association with Alzheimer’s disease risk variants (GWAS), regulatory enhancers, DNA motifs and TF binding sites linked to neuronal differentiation. Large-scale analyses were executed via Nextflow pipelines on Kubernetes and AWS, ensuring scalable and reproducible processing of NGS datasets.
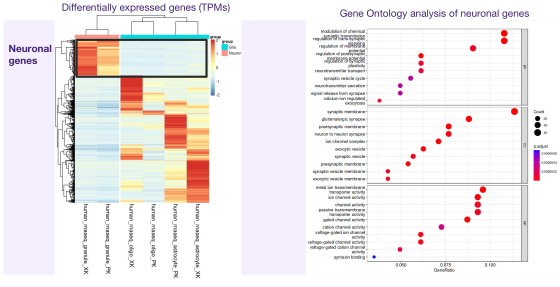
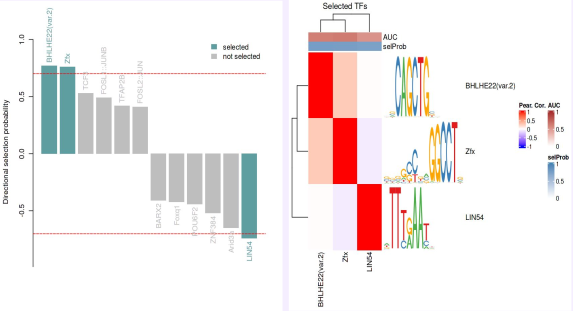
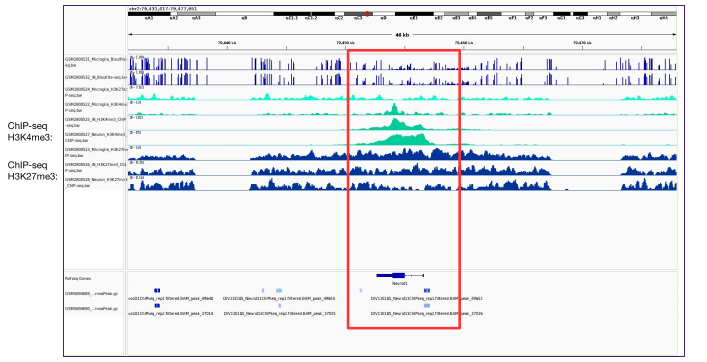
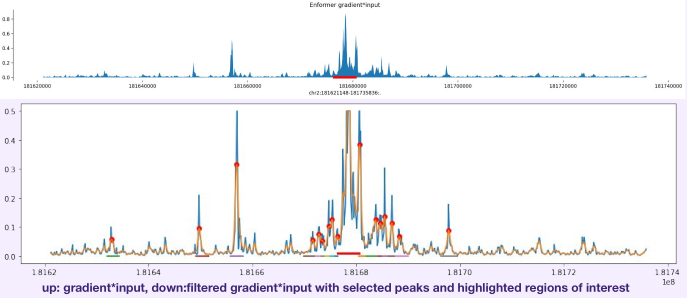
Using DeepMind’s Enformer model by Avsec et al., I mapped the regulatory landscape around the target TF to uncover and confirm DNA regions predicted to most strongly influence its expression in neurons and glia. Enformer predicts regulatory activity directly from DNA sequence and uses gradient-based attribution to reveal regions with the strongest impact on gene expression. I analyzed a ~400 kb region around the transcription factor, applying cell-type–specific masks and signal smoothing to identify candidate enhancers.
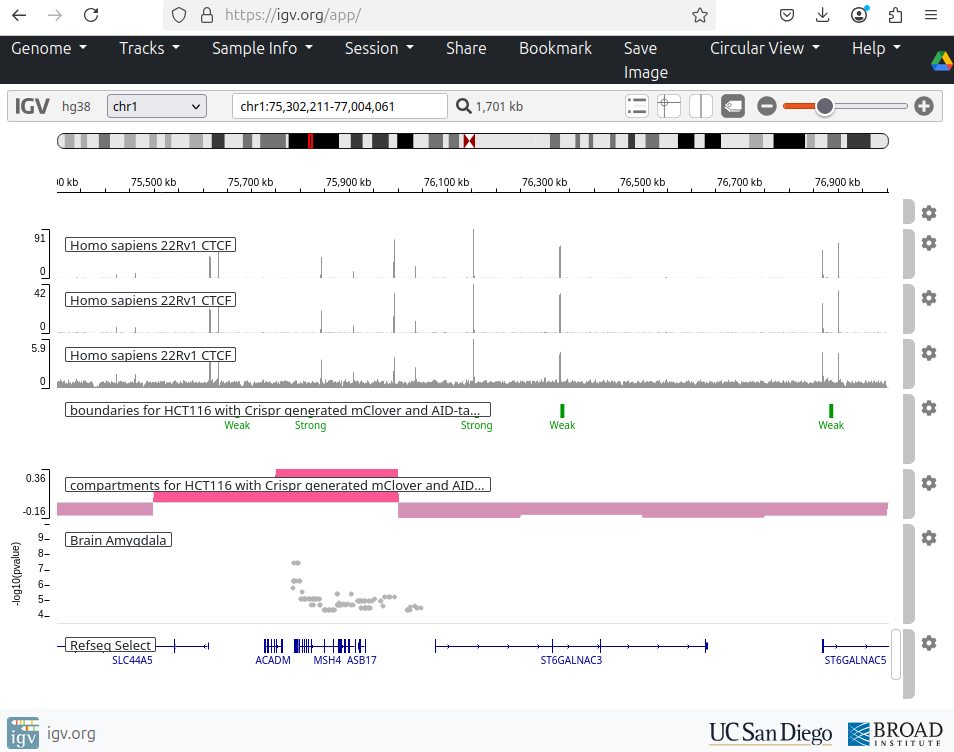
I contributed to enhancing the IGV web application, an interactive tool for visual exploration of genomic data (source code). Built with JavaScript and Python, this tool allows visualization of both public and in-house datasets.
- Enabled dynamic visualization of new in-house genomic datasets.
- Added highlighting of genomic regions of interest (e.g., genetic variants).
- Developed new display options for RefSeq and GENCODE annotations:
- Collapse/expand all transcript isoforms.
- Extend selected gene isoforms for detailed view.
- Added controls to adjust track widths for optimal display.
- Linked visualized tracks to their source databases.
- Implemented command-line tool for automated snapshots of defined genes or regions.
Projects
The aim of this project was to predict gene type directly from a raw DNA sequence using classical machine learning models, convolutional neural networks, and a custom nucleotide-level Transformer, while also exploring practical deployment of machine learning services with Docker compose, Kubernetes (locally with Kind) and cloud infrastructure (AWS Kubernetes EKS)
The goal of this project was simple: Given a set of molecular descriptors, predict whether a compound will dissolve in water. We provide the model with numeric features that describe the molecule (size, polarity, solvation energy, charge distribution, etc.) and ask: "Will this molecule dissolve, or will it stay stubbornly solid?". I experimented with several regression models — Partial Least Squares (baseline linear model), Elastic Net (linear model with feature selection), Random Forest, and XGBoost (gradient-boosted trees) - to predict whether a molecule will dissolve in water based on its descriptors.
The aim of this project was to evaluate transfer learning-based convolutional neural networks for classifying immune cell types from H&E-stained blood microscopy images. I applied transfer learning with a pretrained Xception model as a fixed feature extractor and trained a small custom MLP classification head on top of it. I additionally extracted Xception embeddings and trained classical machine learning models such as logistic regression and XGBoost to provide an interpretable baseline. Hyperparameters such as learning rate and dropout rate were tuned, and the model using the MLP head achieved strong accuracy on the test set.
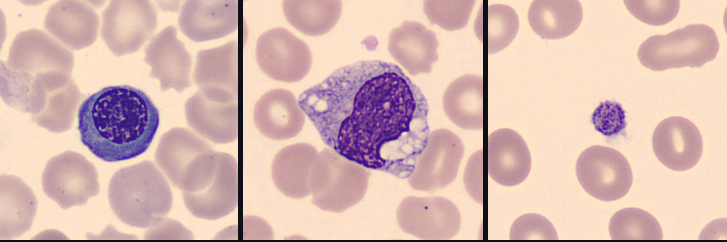
The aim of this project was to explore the ability of diffusion models to generate missing regions in images. It restores corrupted areas in H&E cell images using a compact diffusion model and a simple Streamlit UI.
I developed several survival models to predict the risk of mortality or relapse in newly diagnosed multiple myeloma patients, using baseline clinical and/or gene expression data. The workflow involved RNA-seq preprocessing, unsupervised exploratory analysis (PCA, clustering), and multiple survival modeling strategies - Cox regression, random survival forests, LASSO-based feature selection, and pathway-informed models - evaluated using the C-index.
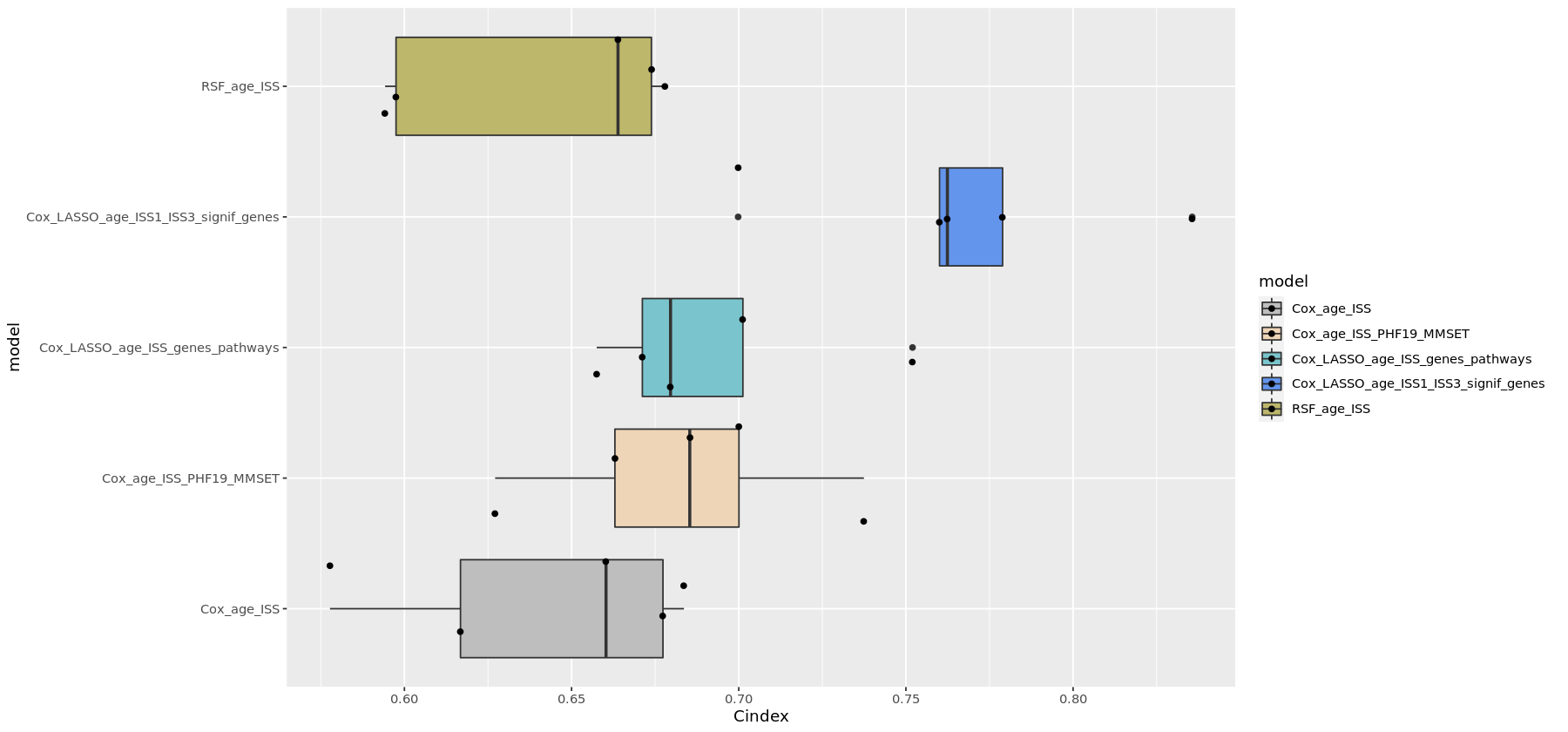
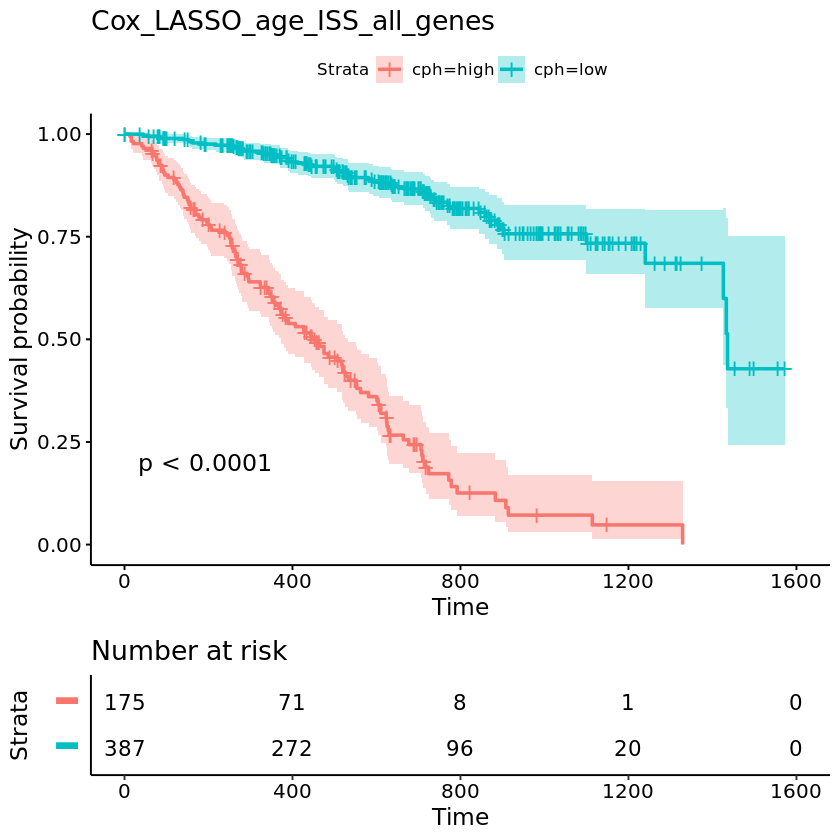
Figure: C-index comparison of multiple survival models (left) and Kaplan–Meier plot of the best performing model (right).
I applied convolutional neural networks (CNNs) to classify chest X-ray images using both 224×224 and 64×64 pixel inputs, aiming to explore whether lightweight models can retain sufficient diagnostic power for image-based classification tasks. In addition to training a baseline CNN from scratch, I employed transfer learning with pretrained convolutional backbones such as ResNet to evaluate whether pretrained models could further enhance classification performance on chest X-ray images.
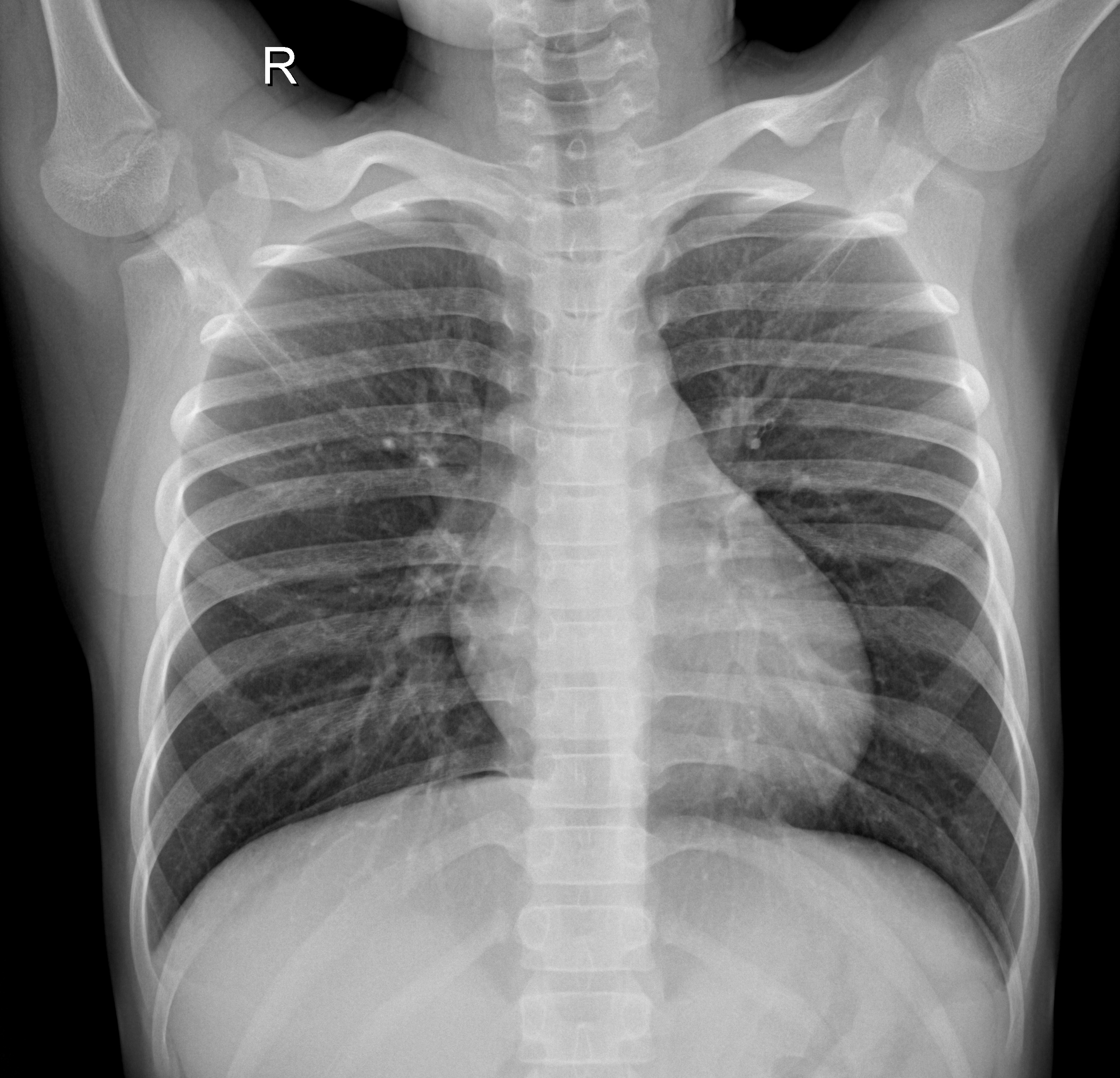
Healthy
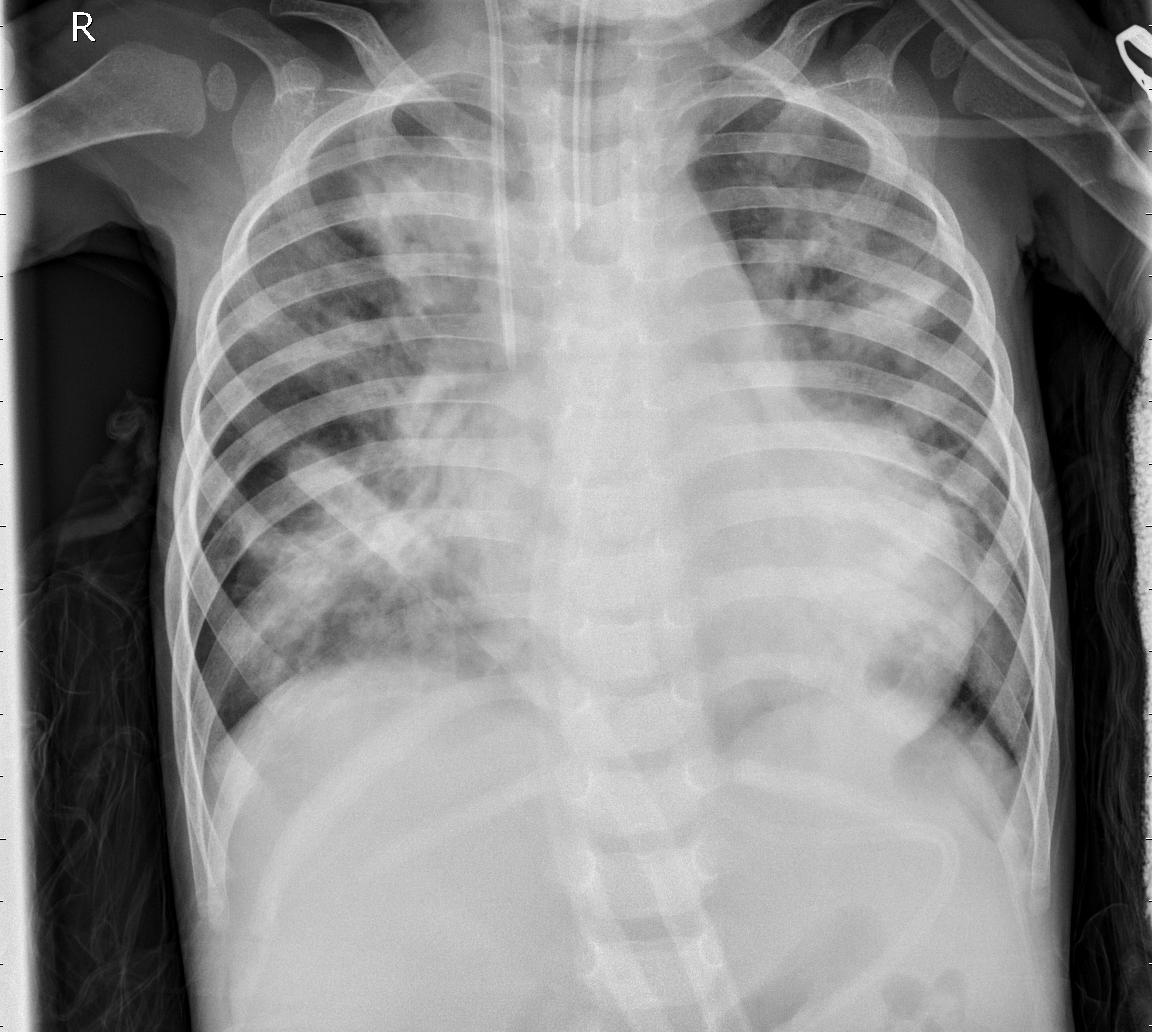
Pneumonia
Figure: Example X-ray images of a healthy individual and a pneumonia patient.
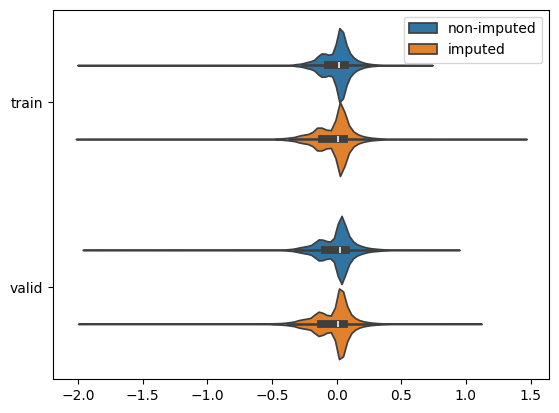
I developed a simple autoencoder with a custom loss function for imputing missing values in single-cell RNA-seq data. The approach was inspired by the method proposed by Badsha et al. (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7144625/).
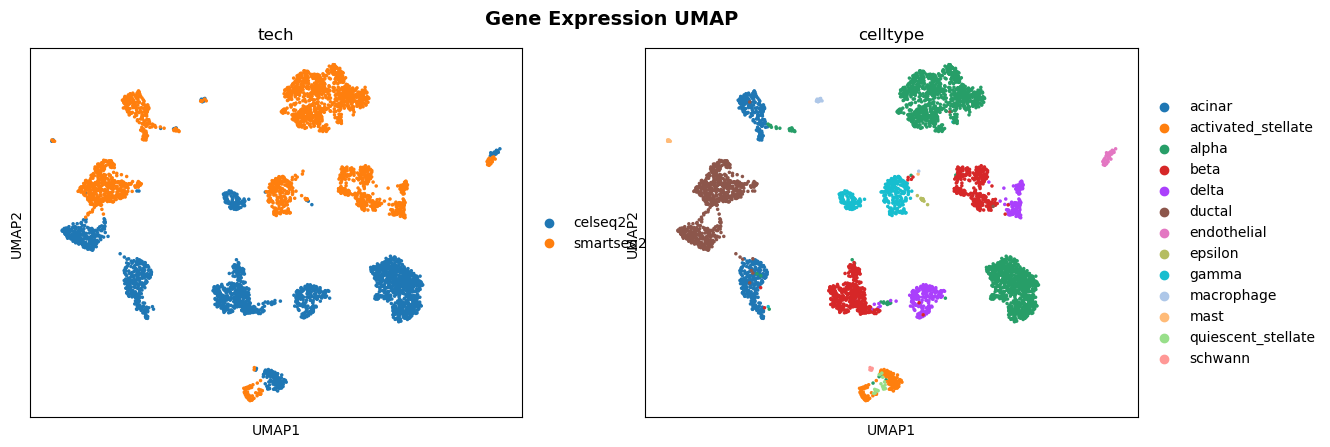
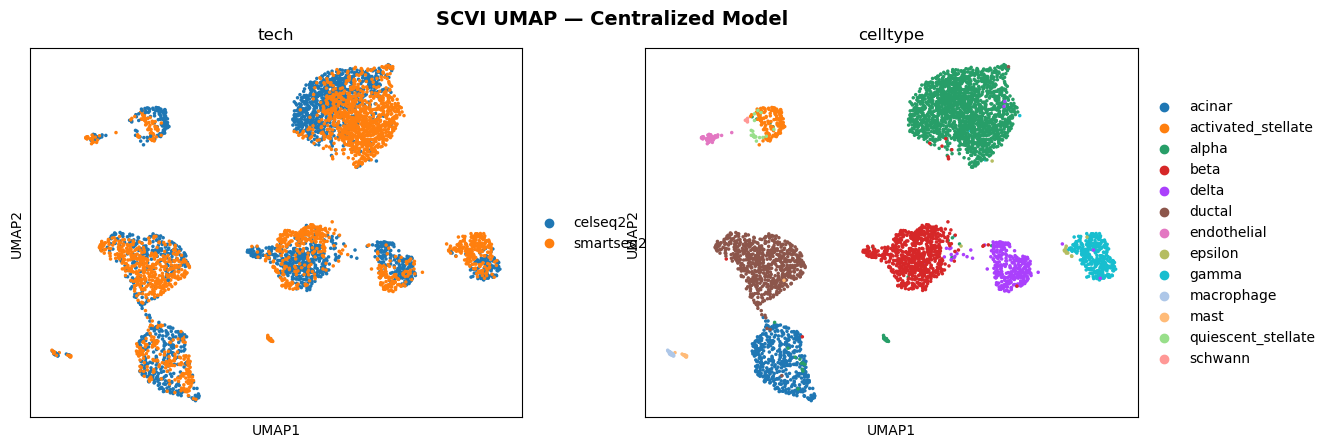
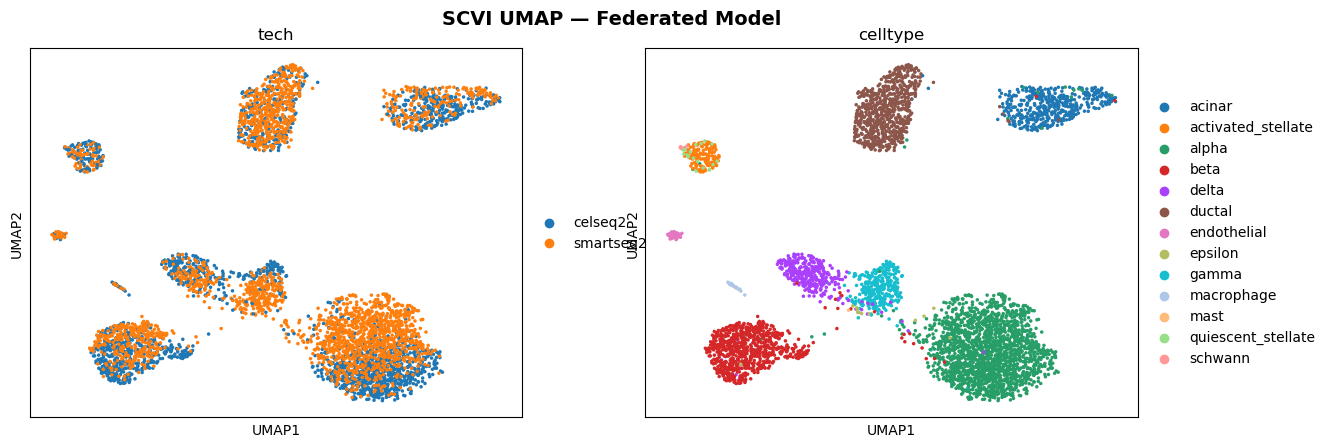
This project explored a scVI model (variational autoencoder for single-cell data) in a federated setting using the Flower framework (Flower.ai) and the SecAgg+ secure aggregation protocol. For comparison, the same model was also trained in a centralized setting.
This project studies cell-free DNA (cfDNA) fragments that circulate in the blood. These fragments originate from many different cell types across the body. When tissues are damaged or diseased, they release more DNA than usual, altering the overall composition of cfDNA in the bloodstream. By identifying which cell types the DNA comes from, we can gain an early view of tissue health and disease signals.
I applied several deconvolution methods to estimate cell type proportions from bulk DNA methylation data. Regression-based approaches (NNLS, Lasso, Ridge, Elastic Net) model methylation profiles as mixtures of reference cell types. In addition, I developed:
- A variational autoencoder (VAE) that reconstructs CpG profiles while jointly predicting cell type proportions.
- A semi-supervised NMF (ssNMF) that anchors factorization to known reference signatures.
- A lightweight transformer model, treating CpG regions as tokens with embeddings and self-attention to capture genomic dependencies.
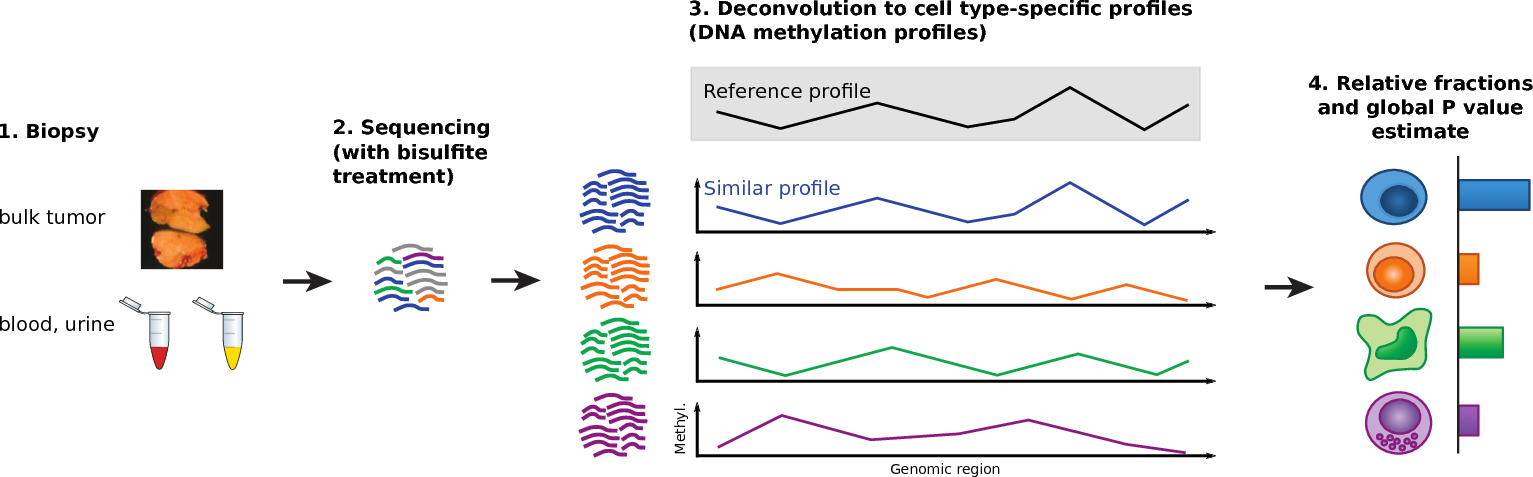
This project's aim was to demonstrate how GNNs can capture spatially coherent patterns in gene expression and to compare these learned embeddings to traditional PCA and k-means-based clustering.
Spatial transcriptomics captures gene expression while preserving tissue architecture, enabling the study of cellular organization and microenvironments. However, identifying coherent spatial domains, regions of similar expression patterns and spatial context, remains challenging.
Graph Neural Networks (GNNs) are great for this type of data because they can model both gene expression features and spatial neighborhood relationships. In this project, I implemented a mini Graph Autoencoder (GAE) from scratch in PyTorch to learn unsupervised spatial embeddings of tissue spots from a toy Visium H&E dataset provided by Squidpy (a 10x Genomics Visium H&E mouse brain section (~2,700 spots, ~33k genes)).
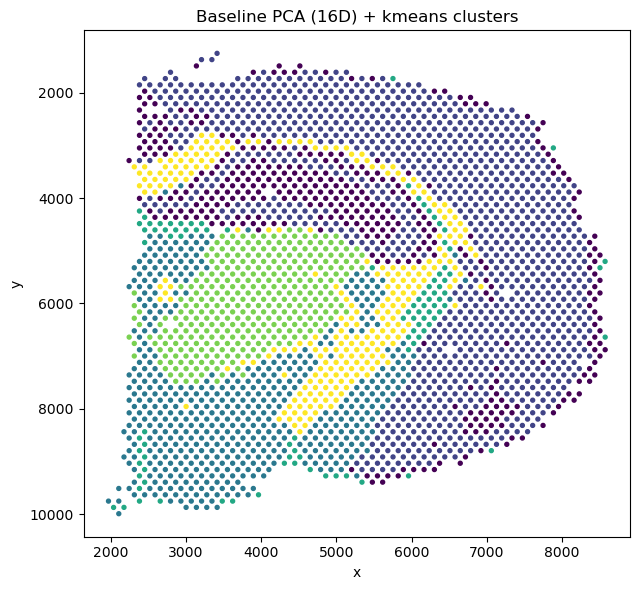
Figure: Baseline PCA + KMeans.
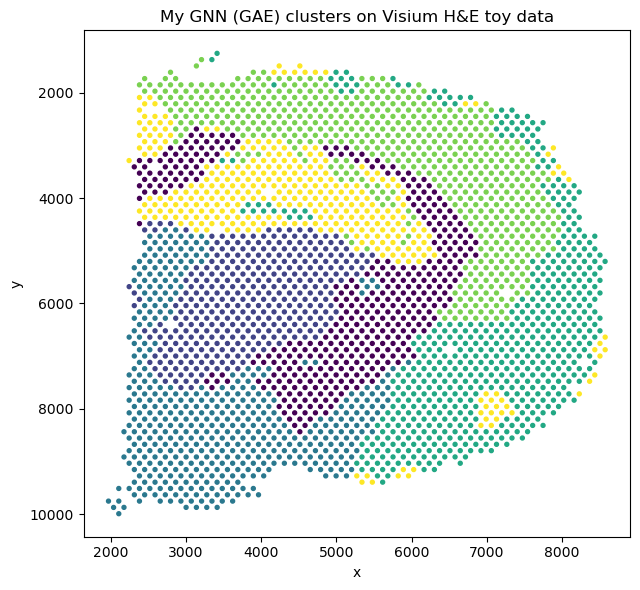
Figure: GNN-based clustering.
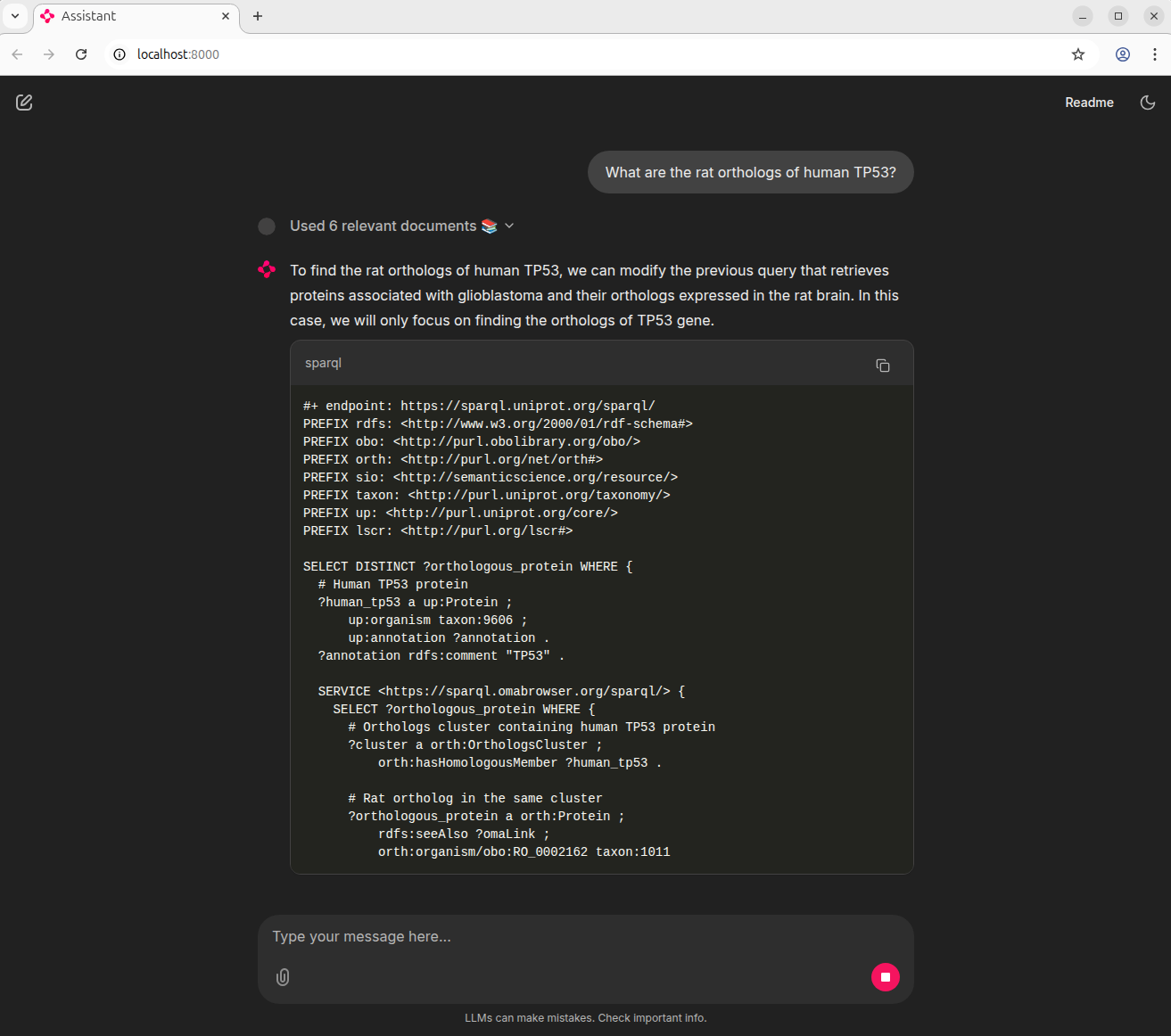
This project explored an AI-powered assistant that helps researchers ask questions about biology in plain English and automatically turns them into SPARQL queries against public databases:
- UniProt - proteins, sequences, and annotations
- OMA - orthologs and evolutionary relationships
- Bgee - gene expression across species
The assistant is powered by LLMs (Mistral, Llama via Groq, Ollama) combined with retrieval-augmented generation (RAG) using Qdrant and FastEmbed. You can interact with the assistant either in the terminal/CLI or through a simple chat web app (Chainlit web UI).
Key goals:
- Allow researchers to query complex biological knowledge bases with a nice web interface.
- Validate and execute queries automatically.
- Provide results summarized in plain language.
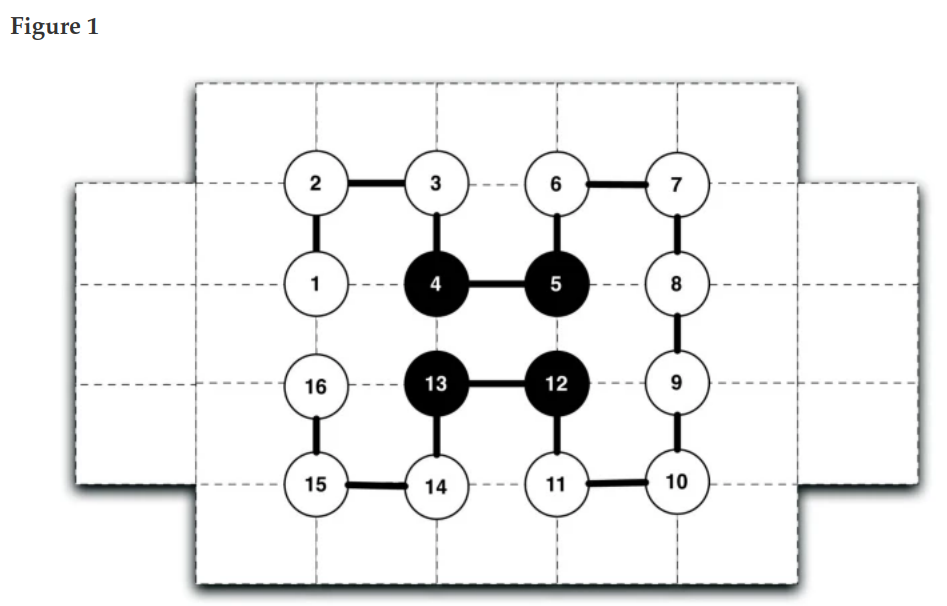
Implementation of simulated annealing and replica exchange Monte Carlo algorithm for protein folding in the Hydrophobic Polar (HP) model in Python and NumPy. The HP model simplifies protein folding by using hydrophobic (H) and polar (P) amino acids on a square lattice. Metropolis–Hastings algorithm enables sampling protein configurations based on the Boltzmann distribution.
Implementation of de Bruijn graph-based genome assembly with Eulerian walk to reconstruct DNA sequences from k-mers. Includes short-read assembly principles based on publications by Compeau et al. (2011) and Pevzner et al. (2001)
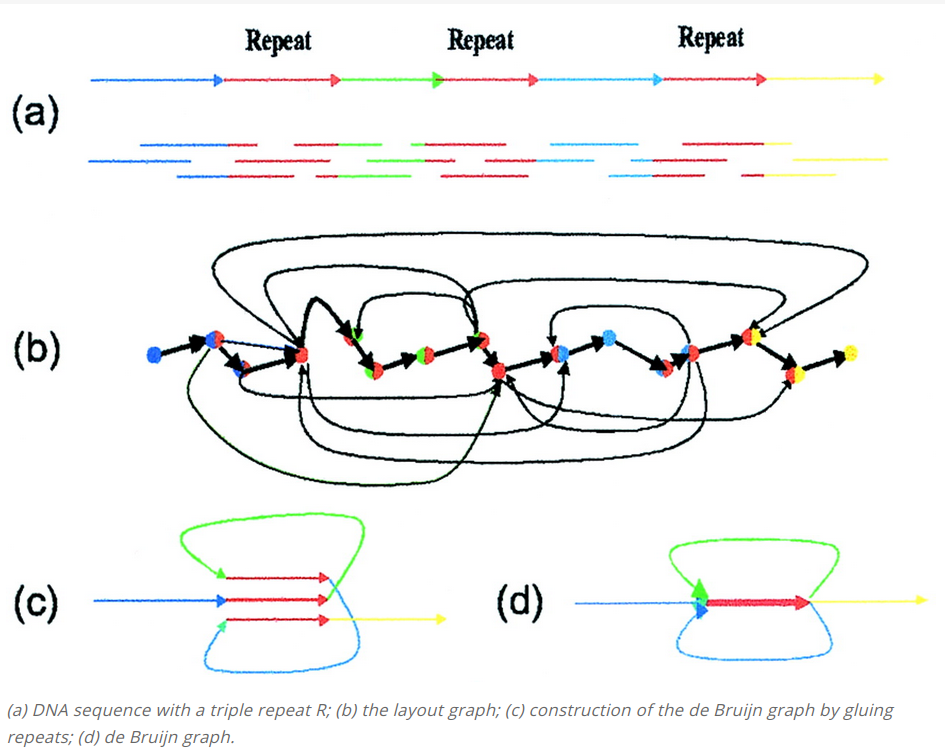
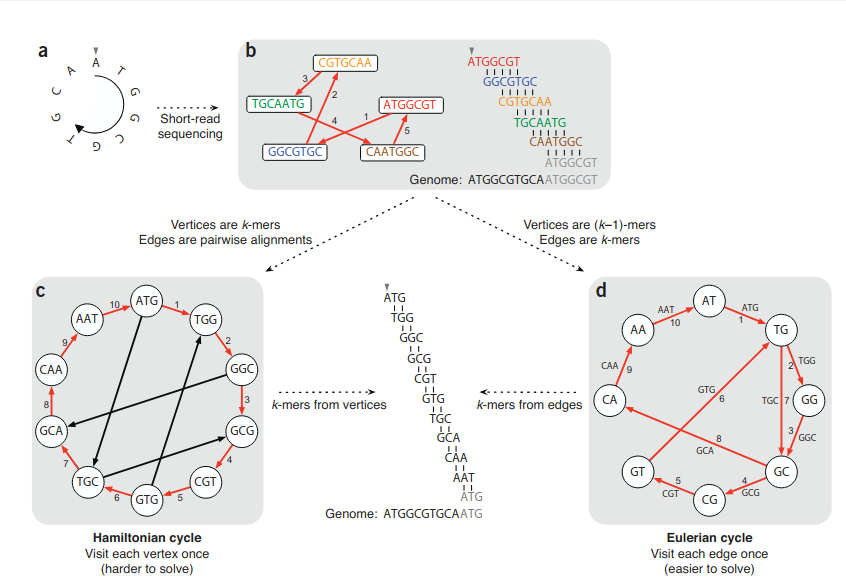
My focus was on modern short-read assembly algorithms construct a de Bruijn graph by representing all k-mer prefixes and suffixes as nodes and then drawing edges that represent k-mers having a particular prefix and suffix. For example, the k-mer edge ATG has prefix AT and suffix TG. Finding an Eulerian cycle allows one to reconstruct the genome by forming an alignment in which each successive k-mer (from successive edges) is shifted by one position. This generates the same cyclic genome sequence without performing the computationally expensive task of finding a Hamiltonian cycle (as shown in the figure below).
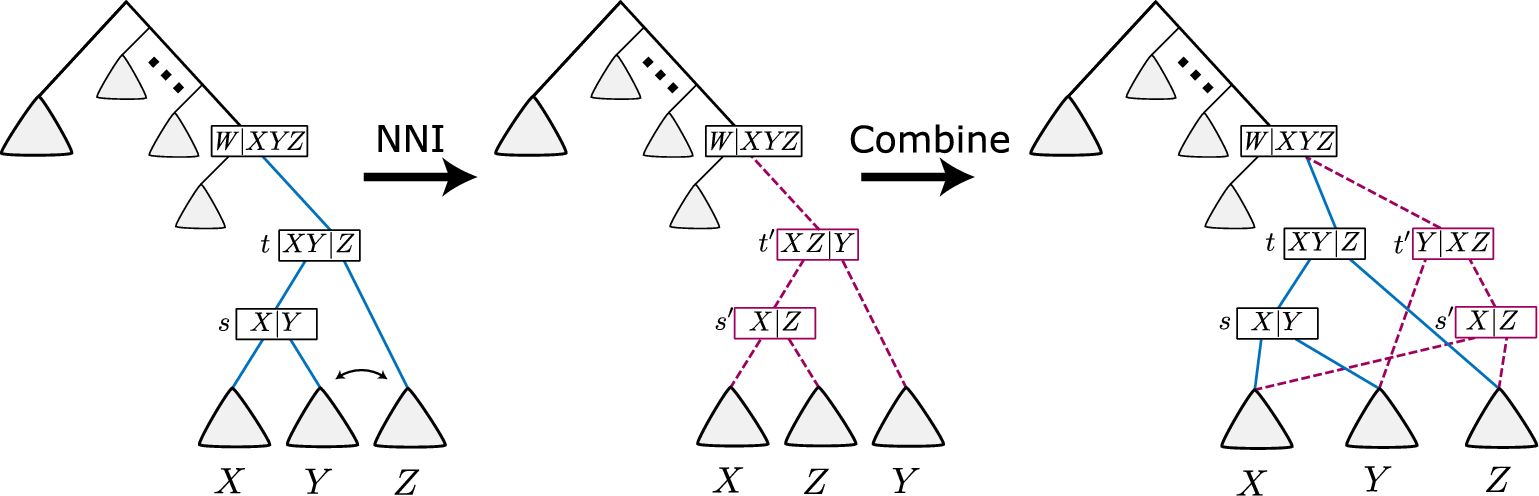
Implementation of the Felsenstein's tree-pruning and the Nearest-Neighbor Interchange (NNI) algorithms. The Felsenstein's tree-pruning is a heuristic algorithm for computing the likelihood of an evolutionary tree from nucleic acid sequence data. It is devoted to searching for an optimal tree structure. NNI is great for rooted binary phylogenetic trees and using Jukes and Cantor substitution model. It is one of the simplest tree-rearrangement methods.
Regulatory DNA discovery - MSA & binomial enrichment
Bio Motif Ensembl is a Python tool for discovering potential regulatory DNA regions across related mammalian genomes using Ensembl’s public MySQL databases. It retrieves orthologous gene sequences (e.g., human, mouse, rat), aligns their upstream regions, and detects conserved non-coding segments. These conserved blocks are then analyzed with motif discovery algorithms such as MEME and AlignACE, and tested for statistical overrepresentation using a binomial model. The framework integrates comparative genomics, multiple sequence alignment, and motif enrichment to identify functionally significant regulatory elements.
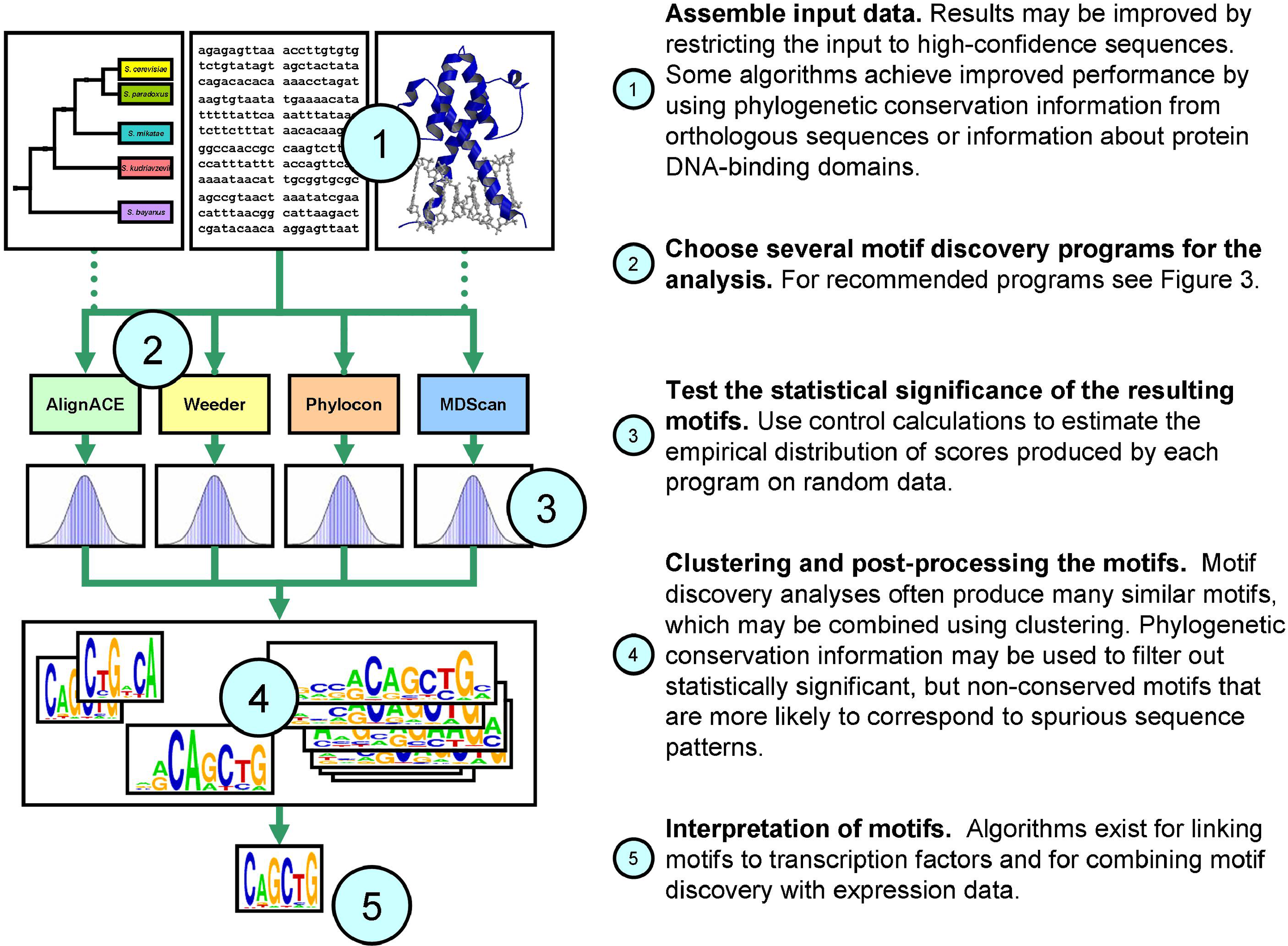
Figure: It's a similar concept to the approach published by MacIsaac et al. 2025.
A simple Sudoku game implemented in JavaScript and JQuery.
A classic Minesweeper game implemented in Java using SWING and AWT libraries.
Django-based server for Multiple Sequence Alignment (MSA) visualization - 🔗 GitHub Repository
Mobile application using Django, manifesto app, and localStorage - 🔗 GitHub Repository
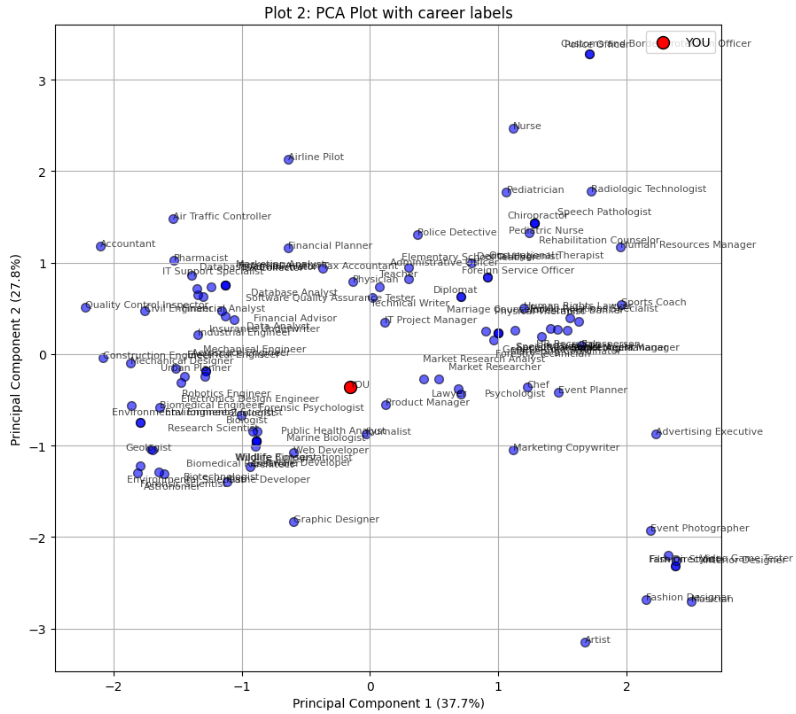
Interactive tool that matches careers to users based on their personality profile (Big Five personality traits). Runs directly in the browser via Pyodide.